Introduction#
Molecular dynamics (MD) simulations are indispensable for gaining atomistic insights into the biomolecular function. MD of increasingly larger proteins has become accessible to researchers with recent advancements in computing and MD algorithms. However, the analysis of MD trajectories remains tedious. MD DaVis (Molecular Dynamics Data Visualizer) is a tool and Python 3 package to perform comparative data analysis of MD trajectories of similar proteins or the same protein under different conditions.
There are many MD analysis tools. However, the output from most has to be visualized using another plotting library requiring a significant amount of coding. The MD DaVis package provides the md-davis command-line tool and md-davis-gui GUI tool to create helpful interactive visualizations easily. The tool can increase the productivity of researchers simulating protein using MD and make the analysis of such simulations accessible to everyone.
Features of MD DaVis#
Click on each panel for more details.
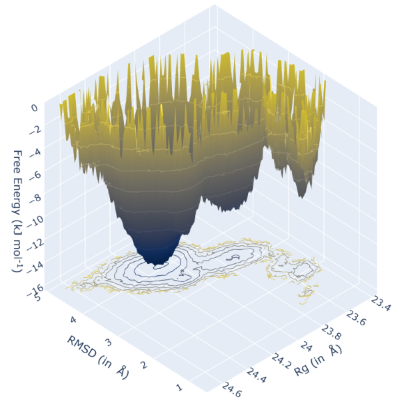
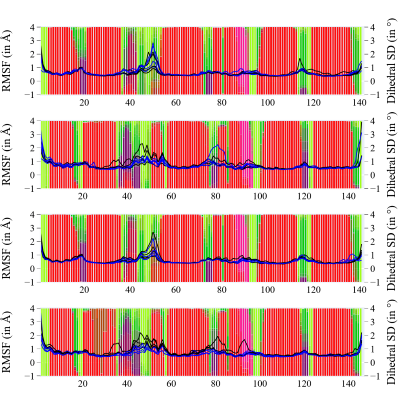
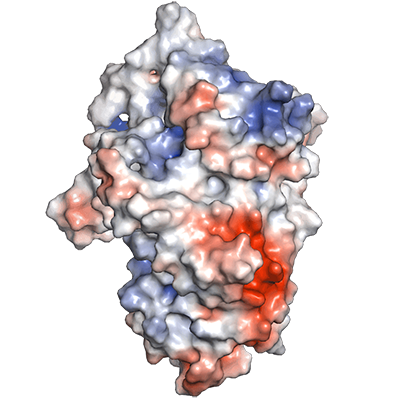
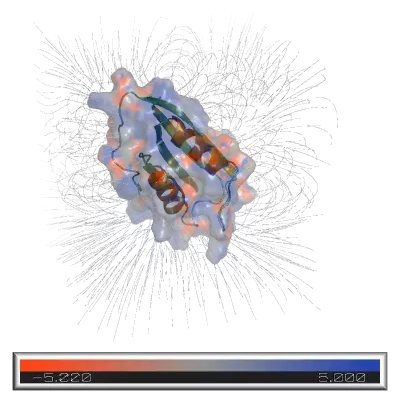
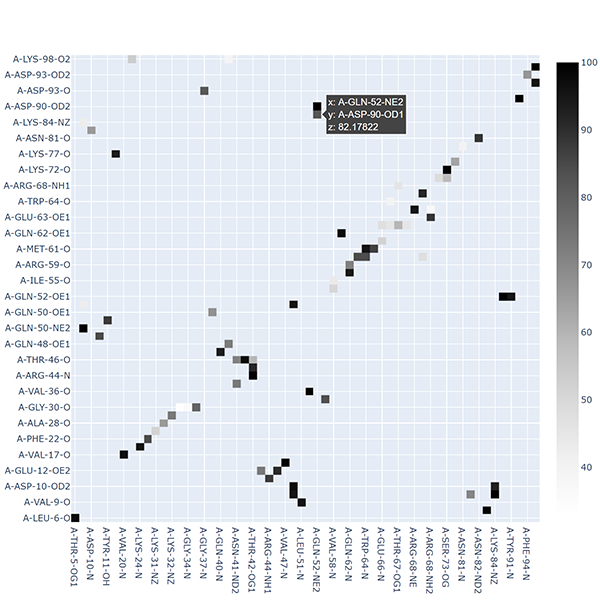
Interfacing MD DaVis to Other Analysis Tools#
MD DaVis does not implement analysis tools and functions available elsewhere. Presently, MD DaVis natively supports analysis performed using GROMACS tools. However, other analysis tools may be used as well, depending on the format of the MD trajectory. The input to most MD DaVis commands is text files with two columns; the first is time or residue number, and the second is the value, e.g., RMSD, Rg, RMSF, SASA.
# gmx rms is part of G R O M A C S:
#
# Gromacs Runs On Most of All Computer Systems
#
@ title "RMSD"
@ xaxis label "Time (ps)"
@ yaxis label "RMSD (nm)"
@TYPE xy
@ subtitle "System after lsq fit to System"
0.0000000 0.0000031
10.0000000 0.0677038
20.0000000 0.0837483
30.0000000 0.0894995
So, interfacing other analysis tools to MD DaVis is as simple as creating these text files, easily accomplished with the Python packages mdtraj or mdanalysis. However, using Python to calculate these properties is significantly slower than using binary tools like those bundled with MD software. Therefore, we recommend using such binary tools whenever possible. A package that can read the trajectory files may be used for the remaining calculations, like mdtraj, which natively accepts binary trajectory files from AMBER, Desmond, GROMACS, LAMMPS, and NAMD. The dihedral angles calculation in MD DaVis is done with mdtraj, and the electrostatics are calculated on a set of PDB files that all MD engines can export. Therefore, MD DaVis does not exclusively rely on GROMACS for these calculations. However, the H-bond/Contact visualization and secondary structure visualization rely on the output from the GROMACS tools hbond and do_dssp, respectively. That is why we recommended converting the trajectory to GROMACS format with mdtraj.
Molecular Dynamics Analysis Tools#
The following is a non-exhaustive list of tools for analysis of MD simulations:
References#
Berendsen, H. J. C. et al. (1995) GROMACS: A message-passing parallel molecular dynamics implementation. Computer Physics Communications, 91, 43-56.
Brehm, M. and Kirchner, B. (2011) TRAVIS - A Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model., 51, 2007-2023.
Brown, D. K. et al. (2017) MD-TASK: a software suite for analyzing molecular dynamics trajectories. Bioinformatics, 33, 2768-2771.
Case, D. A. et al. (2021) Amber 2021 University of California, San Francisco.
Eichenberger, A. P. et al. (2011) GROMOS++ Software for the Analysis of Biomolecular Simulation Trajectories. J. Chem. Theory Comput., 7, 3379-3390.
Grant, B. J. et al. (2006) Bio3d: an R package for the comparative analysis of protein structures. Bioinformatics, 22, 2695-2696.
Margreitter, C. and Oostenbrink, C. (2017) MDplot: Visualise Molecular Dynamics. The R Journal, 9, 164-186.
McGibbon, R. T. et al. (2015) MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys J, 109, 1528-1532.
Michaud-Agrawal, N. et al. (2011) MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. , 32, 2319-2327.
Popov, A. V. et al. (2013) MDTRA: A molecular dynamics trajectory analyzer with a graphical user interface. Journal of Computational Chemistry, 34, 319-325.
Roe, D. R., and Cheatham, T. E. (2013) PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput., 9, 3084-3095.